
Priority research lines of the UETEM
LINES OF RESEARCH OF THE UNIT OF LIPODISTROFIAS (UETeM) OF THE CENTER OF RESEARCH IN MOLECULAR MEDICINE AND CHRONIC DISEASES (CIMUS) OF THE UNIVERSITY OF SANTIAGO DE COMPOSTELA (2003-2017).
 The UETeM was created in 2003 under the auspices of Professors Lado-Abeal and Araújo-Vilar in the Faculty of Medicine of Santiago de Compostela. Since 2008, the unit has been dedicated exclusively to research on the clinical and molecular aspects of infrequent lipodystrophic syndromes. Since this year, its director is Dr. David Araújo-Vilar, Professor of Medicine and specialist in Endocrinology and Nutrition of the University Hospital of Santiago de Compostela (CHUS) (Figure 1), responsible for the service Lipodystrophy Unit of Endocrinology of CHUS, which is integrated into the National Reference Center (CSUR) of Hereditary Metabolic Diseases and European Expert Center (MetabERN). At the same time, Dr. Araújo-Vilar is one of the founders in 2012 of the European Lipodystrophy Consortium (ECLip, www.european-lipodystrophies.org/en). Since 2012, the laboratories of the UETeM are located in the Center for Research in Molecular Medicine and Chronic Diseases (CiMUS) of the University of Santiago de Compostela (Figure 2).
The UETeM was created in 2003 under the auspices of Professors Lado-Abeal and Araújo-Vilar in the Faculty of Medicine of Santiago de Compostela. Since 2008, the unit has been dedicated exclusively to research on the clinical and molecular aspects of infrequent lipodystrophic syndromes. Since this year, its director is Dr. David Araújo-Vilar, Professor of Medicine and specialist in Endocrinology and Nutrition of the University Hospital of Santiago de Compostela (CHUS) (Figure 1), responsible for the service Lipodystrophy Unit of Endocrinology of CHUS, which is integrated into the National Reference Center (CSUR) of Hereditary Metabolic Diseases and European Expert Center (MetabERN). At the same time, Dr. Araújo-Vilar is one of the founders in 2012 of the European Lipodystrophy Consortium (ECLip, www.european-lipodystrophies.org/en). Since 2012, the laboratories of the UETeM are located in the Center for Research in Molecular Medicine and Chronic Diseases (CiMUS) of the University of Santiago de Compostela (Figure 2).

The UETeM is currently constituted by the following members: David Araujo-Vilar (Endocrinologist and Professor of Medicine), Joaquín Lado-Abeal (Endocrinologist and Associate Professor of Medicine, accredited to the University Professor by the ANECA), Sofía Sánchez- Iglesias (PhD in Biochemistry and responsible for laboratory), Cristina Guillín-Amarelle (endocrinologist and doctor of medicine), Antía Fernández-Pombo (resident doctor of Endocrinology and Nutrition and PhD student) and Yanis Bouzaher (graduate in Biology and PhD student ), also collaborating with medical students (María González-Alonso, Lia García-Formoso and Zeus Díaz-Vilela) (Figure 3 and 4).


Research lines 1.
Celia encephalopathy
1. The disease: This childhood neurodegenerative disease was discovered at the UETeM in 2011 by Dr Araújo-Vilar (Guillén-Navarro et al J Med Genet 2013). It is a very infrequent disorder (less than one case per 1,000,000,000 people) characterized by an extremely severe encephalopathy that begins in early childhood. Over 3 years, these children suffer a rapid and progressive neurological involution that causes them to lose their brain functions (language, motor functions, interaction with the environment), appearing a myoclonic epilepsy of very difficult control, and which leads to death before 9 years old In some cases (vide infra), children also present at a very young age (months) a loss of generalized fat loss (l ipodist rofia), which leads to metabolic and hepatic complications (hypertriglyceridemia, diabetes, fatty liver). To date, 7 cases of the disease have been described, of which 6 have died. This disorder is a very serious variant of generalized congenital lipodystrophy type 2 (Berardinelli-Seip type 2 syndrome). Recently, our group has identified a variant of Celia encephalopathy due to a different mutation in the BSCL2 gene (vide infra), with a practically superimposable clinical picture but with a somewhat longer survival (10-11 years) (unpublished data)
2. The cause: Celia encephalopathy, also called PELD (Progressive Encephalopathy with or without lipodystrophy, MIM: # 615924, https://www.omim.org/entry/ 615924 ? Search = peld & highlight = peld) is caused by the mutation c.985C> T in the BSCL2 gene, located on chromosome 11. It is a recessive disorder, that is, it is necessary that both alleles of the gene are affected so that the disease develops, being the parents asymptomatic carriers. Patients who are homozygous (ie, those who have the mutation c.985C> T in the maternal and paternal alleles) do not exhibit adipose tissue involvement, however the children are heterozygous compounds (the mutation c.985C> T is present in one of the alleles, and another different mutation in the same gene is in the other), in addition to the neurodegenerative disease they present generalized lipodystrophy. (Figure 5)
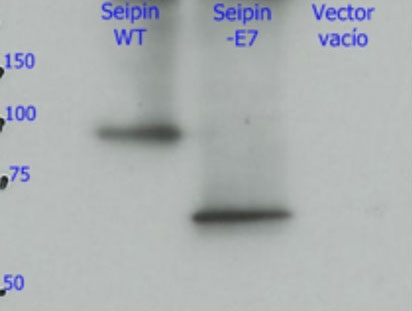
3. The mechanisms: Why does this mutation cause such serious damage in the brain? Studies carried out in our laboratory (Guillén-Navarro et al J Med Genet 2013, Ruiz-Riquelme et al Neurobiol Dis 2015) have allowed to reveal why the mutation c.985C> T damages the brains of these children so dramatically. This small change in exon 7 of the BSCL2 gene causes the disappearance thereof (of exon 7) during transcription (the passage of DNA to RNA). The loss of this region of the BSCL2 gene causes the protein that is translated from the RNA (seipine) to be an abnormal or aberrant protein (we will call it seipina-Celia) with a tendency to form large aggregates of seipina-celia both in the cytoplasm as the nucleus of neurons. These macro-aggregates of seipina-Celia can not be eliminated by the defense mechanisms of the cells when they encounter "diseased" proteins (what is called UPR, response to misfolded proteins), which produces stress of the endoplasmic reticulum, and at one point, apoptosis (programmed cell death). In other words, as the months pass, in the brain neurons of these children accumulate large amounts of sepia-Celia which is "toxic" to them, so they die. The constant loss of neurons explains the progressive and lethal neurological picture. ( figure 6)
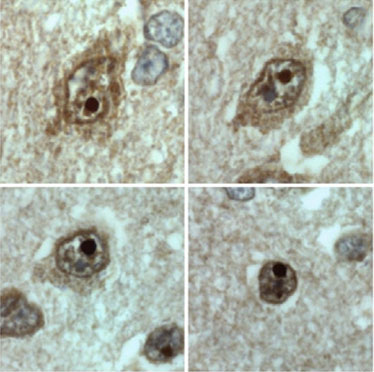
4. The treatment: To this day there is no cure for this disease. However, our group has made significant progress in slowing neurological damage. For 4 and a half years we have been treating the only living girl with PELD with recombinant human leptjna and a diet rich in polyunsaturated fatty acids and supplemented with omega-3 fatty acids. Today, this girl, who already has 8 years, is not only alive but has experienced a modest improvement in psychometric tests and, strikingly, in neuroimaging studies (Araujo-Vilar et al EJHG 2017, pending acceptance). Our studies in neuronal models in vitro have confirmed that the association of leptin with docosahexaenoic acid reduces the expression of toxic seipin by 30%. These results, although hopeful, should be taken with great caution since it is a single case. In these moments, we are evaluating other therapeutic options but to cure, at least to delay the process.
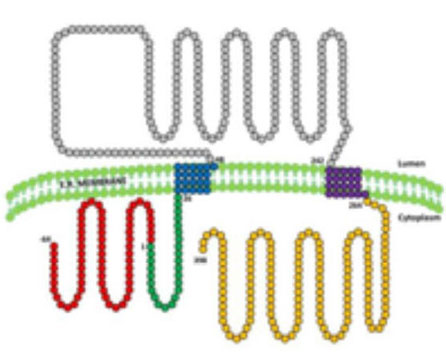 5. What is seipina good for ?: Seipin is a protein resident in the endoplasmic reticulum of which 3 isoforms (variants) are known (figure 8). Studies of our group and others have shown that the longest isoform is expressed mostly in the central nervous system, while in the other tissues it is the isoform of intermediate size that predominates. It is very likely that this protein has different functions depending on the tissue in which it is expressed. The function that has been best studied is related to the formation of lipid drops in the adipose tissue, so that the absence of seipine or the synthesis of a defective seipine would alter the formation of these drops that are crucial in the development of adipocytes. . But in addition, there are data that suggest that seipin also plays a role in neurodevelopment and our group has found evidence to suggest that this protein could play a protective role against oxidative stress in neurons (article in preparation) (Figure 9).
5. What is seipina good for ?: Seipin is a protein resident in the endoplasmic reticulum of which 3 isoforms (variants) are known (figure 8). Studies of our group and others have shown that the longest isoform is expressed mostly in the central nervous system, while in the other tissues it is the isoform of intermediate size that predominates. It is very likely that this protein has different functions depending on the tissue in which it is expressed. The function that has been best studied is related to the formation of lipid drops in the adipose tissue, so that the absence of seipine or the synthesis of a defective seipine would alter the formation of these drops that are crucial in the development of adipocytes. . But in addition, there are data that suggest that seipin also plays a role in neurodevelopment and our group has found evidence to suggest that this protein could play a protective role against oxidative stress in neurons (article in preparation) (Figure 9).
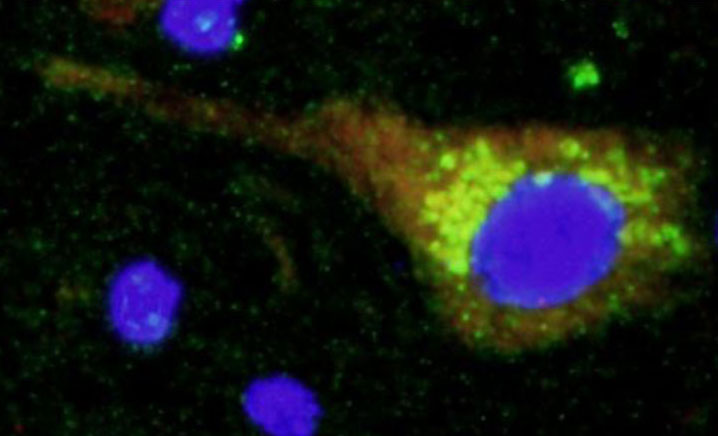
2. Search for new genes involved in familial partial lipodystrophy.Familial partial lipodystrophy is a Mendelian disease that can be caused by mutations in different genes. To date, 7 genes have been identified, but in more than 50% of patients it has not been possible to find mutations in them. One of the lines of research of our group in finding new genes responsible for this subtype of lipodystrophies. Although today we have located a couple of new candidate genes (articles in preparation), our most ambitious project in this field is being carried out in collaboration with the University of Cambridge and the Welcome Trust Sanger Institute in the United Kingdom. With them we are analyzing the genome of more than 60 patients with familial partial lipodystrophy type 1.
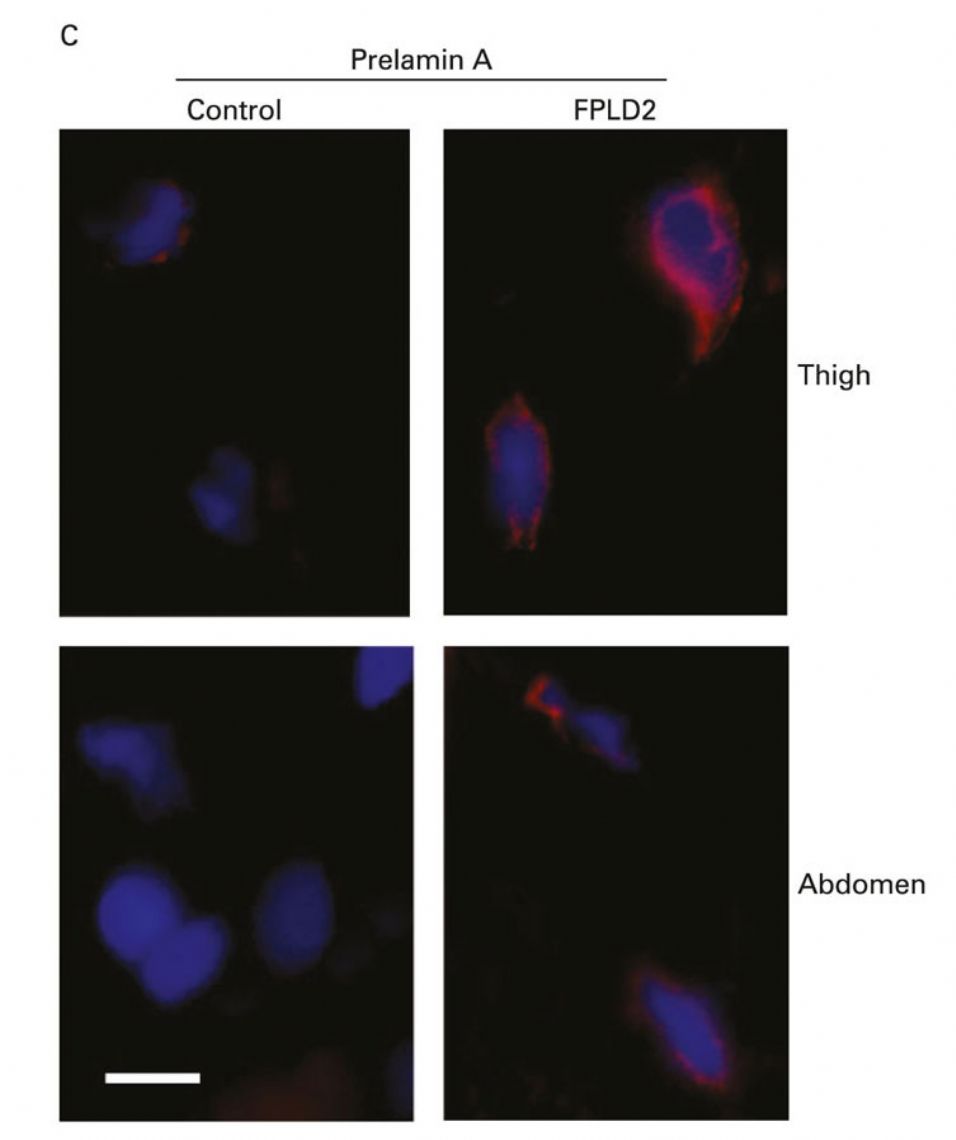 3. Pathogenetic mechanisms of Dunnigan's disease. Dunnigan's disease or Family Partial Lipodystrophy type 2 is caused by certain mutations in the LMNA gene, a gene that encodes a nuclear protein called Lamin A / C. To this day it is not known with certainty through which mechanisms these mutations give rise to such a particular loss of adipose tissue. Studies carried out in our laboratory and by other groups suggest that certain mutations in LMNA prevent an adequate processing of Lamin A / C leading to an accumulation of the immature form of the protein, prelamin A. This excess of immature protein would alter the processes of adipocyte differentiation (Araujo-Vilar et al., J Med Genet 2009) (Figure 10). However, this mechanism would be exclusive of adipocytes, because in fibroblasts of these patients such accumulation does not take place (Y Tu et al., Nucleus 2016) (figure 11). Recently, in a collaboration with Giovanna Lattanzi in Bologna, we have shown that the accumulation of prelamin A alters the autophagic processes, both in white fat and in brown fat in patients with this disease (C Pellegrini et al., Cell Death Differ. , pending acceptance). However, recently, a work by the Philippe Collas group in Norway has shown the importance of the alteration of certain epigenetic mechanisms in the failure of adipogenesis in these patients. In the coming months we intend to initiate a series of experiments in preadipocytes of patients with this disease to deepen the epigenetic alterations.
3. Pathogenetic mechanisms of Dunnigan's disease. Dunnigan's disease or Family Partial Lipodystrophy type 2 is caused by certain mutations in the LMNA gene, a gene that encodes a nuclear protein called Lamin A / C. To this day it is not known with certainty through which mechanisms these mutations give rise to such a particular loss of adipose tissue. Studies carried out in our laboratory and by other groups suggest that certain mutations in LMNA prevent an adequate processing of Lamin A / C leading to an accumulation of the immature form of the protein, prelamin A. This excess of immature protein would alter the processes of adipocyte differentiation (Araujo-Vilar et al., J Med Genet 2009) (Figure 10). However, this mechanism would be exclusive of adipocytes, because in fibroblasts of these patients such accumulation does not take place (Y Tu et al., Nucleus 2016) (figure 11). Recently, in a collaboration with Giovanna Lattanzi in Bologna, we have shown that the accumulation of prelamin A alters the autophagic processes, both in white fat and in brown fat in patients with this disease (C Pellegrini et al., Cell Death Differ. , pending acceptance). However, recently, a work by the Philippe Collas group in Norway has shown the importance of the alteration of certain epigenetic mechanisms in the failure of adipogenesis in these patients. In the coming months we intend to initiate a series of experiments in preadipocytes of patients with this disease to deepen the epigenetic alterations.
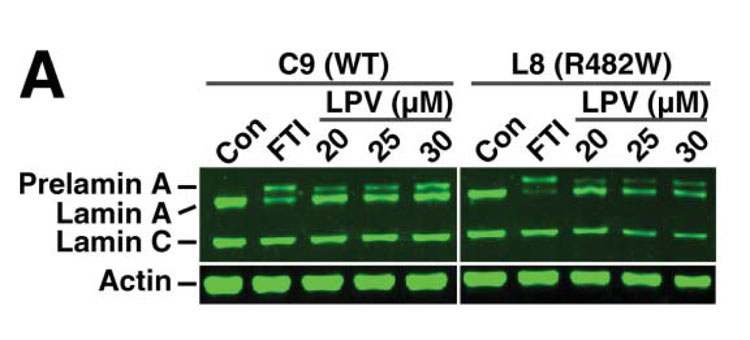
4. New treatments for infrequent lipodystrophies
1. Human recombinant leptin: Today, in addition to diet and physical exercise, treatment with human recombinant leptin is the only one that allows adequate control of metabolic and liver complications associated with generalized lipodystrophy. The available data on its efficacy in familial partial lipodystrophy suggest that it may be beneficial in certain patients. Leptin does not allow the restitution of adipose tissue. Human recombinant leptin (metreleptin) has been approved by the health authorities in the USA and Japan. In some European countries, including Spain, it has been prescribed as compassionate use for 10 years. Our group in Santiago de Compostela is the only one in Spain that has been authorized for its prescription. As of today we are treating 11 patients with this drug, and we are waiting for authorization for three more patients (Figure 12).
2. Other therapeutic alternatives:
1. Our group participates in a multicenter clinical trial in phase 2/3 where the efficacy of a new treatment of hypertriglyceridemia in familial partial lipodystrophy is being tested. It is a drug of the second generation antisense oligonucleotide type that hybridizes with the messenger RNA of the apoprotein apoprotein-III gene.
2. Simultaneously, based on ex vivo and in vitro experiments carried out in our laboratory, we have certain evidences that make us optimistic about certain drugs that are already being used for other pathologies (therapeutic repositioning) and that could be useful for at least slow down the neurodegenerative process of Celia encephalopathy. We hope that with the development of our transgenic murine model we can have even more solid data in this regard.
 5. European Lipodystrophy Registry: After two years of scientific discussions and discussions, the members of the European Lipodystrophy Consortium (ECLip) (http:// www.european-lipodystrophies.org/en/) we have approved the development of a European registry of patients with infrequent lipodystrophies (http://134.60.15.143:8080/login.xhtml) (Figure 13). The register will be based at the German University of Ulm and will be financed with funds from ECLip members. The intention is to maintain the registry for at least 10 years and be able to carry out a detailed study of the natural history of the different subtypes of lipodystrophies (figure 14). In this sense, the participation of patients and relatives will be critical. According to our forecasts we expect to start the collection of patient data in November 2017.
5. European Lipodystrophy Registry: After two years of scientific discussions and discussions, the members of the European Lipodystrophy Consortium (ECLip) (http:// www.european-lipodystrophies.org/en/) we have approved the development of a European registry of patients with infrequent lipodystrophies (http://134.60.15.143:8080/login.xhtml) (Figure 13). The register will be based at the German University of Ulm and will be financed with funds from ECLip members. The intention is to maintain the registry for at least 10 years and be able to carry out a detailed study of the natural history of the different subtypes of lipodystrophies (figure 14). In this sense, the participation of patients and relatives will be critical. According to our forecasts we expect to start the collection of patient data in November 2017.

Scientific production (2003-2017)
1. Antía Fernández-Pombo, Pablo San Millán-Tejedor, Cristina Guillín-Amarelle, Ana I. Castro, Juan Carlos Guinarte-Cabada, Margarita Ventura-Victoria, David Araújo-Vilar. Morbid obesity and psychiatric disorders hiding a sex aneuploidy: a case of late diagnosis of 48,XXYY syndrome. Journal of Genetic Disorders & Genetic Reports, 2017 (pending acceptance).
2. David Araújo-Vilar, Rosario Domingo-Jiménez, Álvaro Ruibal, Pablo Aguiar, Salvador Ibáñez-Micó, Miguel Garrido-Pumar, Miguel Ángel Martínez-Olmos, Concepción López-Soler, Cristina Guillín- Amarelle, María González-Rodríguez, Antonio Rodríguez-Núñez, Julián Álvarez-Escudero, Mercedes Liñares-Paz, Blanca González-Méndez, Silvia Rodríguez-García, Sofía Sánchez-Iglesias.The effects of metreleptin treatment and dietary intervention on neurological regression in Celia's encephalopathy. European J Human Genet 2017 (pending acceptance).
3. Camilla Pellegrini, Marta Columbaro, Sabino Prencipe, Davide Andrenacci, Laura Zanotti, Patricia Iozzo, Maria Angela Guzzardi, Cristina Capanni, Manuela Loi, Elisa Schena, David Araujo Vilar, Saverio Cinti, Paolo Morselli, Alessandra Gambineri, Giovanna Lattanzi. Altered adipocyte differentiation in FPLD2 is associated with unbalanced autophagy. In vitro and in vivo study of adipose tissue browning. Cell Death & Differentiation, 2017 (pending acceptance).
4. Lado-Abeal J, Martinez-Sanchez N, Cocho JA, Martin-Pastor M, Castro-Piedras I, Saha AK, Lopez M. Septic shock effects on pig myocardial metabolites: a metabolomic approach. Scientific Report. 2017(pending acceptance)
5. Cristina Guillín-Amarelle, Sofía Sánchez-Iglesias, Antonio Mera, Elena Pintos, Ana Castro-Pais, Leticia Rodríguez-Cañete, Julio Pardo, Felipe F. Casanueva, David Araújo-Vilar. Inflammatory myopathy in the context of a bizarre overlapping laminopathy. Archives of Endocrinology and Metabolism, 2017 (pending acceptance)
6. Antía Fernández-Pombo, Javier A. Ossandon-Otero, Cristina Guillín-Amarelle, Sofía Sánchez-Iglesias, Ana I. Castro, Blanca González-Méndez, Silvia Rodríguez-García, Leticia Rodriguez-Cañete, Felipe F. Casanueva, David Araújo-Vilar. Bone mineral density in familial partial lipodystrophy. Clinical Endocrinology, 2017 (pending acceptance).
7. Marta Giralt, Francesc Villarroya, David Araújo-Vilar. Lipodystrophies. Encyclopedia of Endocrine Diseases 2nd Edition, Elsevier 2017 (en prensa).
8. Chiquette E, Oral EA, Garg A, Araujo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 2017 (in press).
9. Jéru I, Vatier C, Araújo-Vilar D, Vigouroux C, Lascols O. Clinical Utility Gene Card for: Familial partial lipodystrophy. Eur J Hum Genet. 2017 Feb;25(2). doi: 10.1038/ejhg.2016.102.
10. Yiping Tu, Sofía Sánchez-Iglesias, David Araújo-Vilar, Loren G. Fong, Stephen G. Young. LMNA missense mutations causing familial partial lipodystrophy do not lead to an accumulation of prelamin A. Nucleus 2016 Sep 2;7(5):512-521.
11. Glynn N, Kenny H, Quisenberry L, Halsall DJ, Cook P, Tun T, McDermott J, Smith D, Thompson CJ, O'Gorman D, Boelen A, Lado-Abeal J, Agha A.. The effect of growth hormone replacement on the thyroid axis in patients with hypopituitarism: in vivo and ex vivo studies. Clinical Endocrinology. 86:747-754. 2017
12. Araújo-Vilar D, Barreiro J, Sánchez-Iglesias S, Guillín-Amarelle C. Acantosis nigricans in severe insulin resistance syndromes. An Pediatr (Barc). 2017 Mar;86(3):166-168. doi: 10.1016/j.anpedi.2016.01.003.
13. RJ Brown, D Araújo-Vilar, PT Cheung, D Dunger, A Garg, M Jack, L Mungai, EA Oral, N Patni, K Rother, J von Schnurbein, E Sorkina, T Stanley, C Vigouroux, M Wabitsch, R Williams, T Yorifuji. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab 2016 Dec;101(12):4500-4511.
14. Panikkath R, Panikkath D, Sánchez-Iglesias S, Araújo-Vilar D, Lado-Abeal J. An Uncommon Association of Familial Partial Lipodystrophy, Dilated Cardiomyopathy, and Conduction System Disease. J Investig Med High Impact Case Rep. 2016 Jul 15;4(3):2324709616658495.
15. Jéru I, Vatier C, Araújo-Vilar D, Vigouroux C, Lascols O. Clinical Utility Gene Card for: Congenital Generalized Lipodystrophy. Eur J Hum Genet. 2016 Nov;24(11). doi: 10.1038/ejhg.2016.53
16. C Guillín-Amarelle, S Sánchez-Iglesias, A Castro-Pais, L Rodriguez-Cañete, L Ordóñez-Mayán, Marcos Pazos, B González-Méndez, S Rodríguez-García, F.F. Casanueva, A Fernández-Marmiesse, D Araújo- Vilar. Type 1 Familial Partial Lipodystrophy: Understanding the Köbberling Syndrome. Endocrine, 2016 Nov;54(2):411-421, DOI: 10.1007/s12020-016-1002-x.
17. Sofía Sánchez-Iglesias, Alexander Unruh-Pinheiro, Cristina Guillín-Amarelle, Blanca González-Méndez, Alejandro Ruiz-Riquelme, Leticia Rodríguez-Cañete, Silvia Rodríguez-García, Encarnación Guillén- Navarro, Rosario Domingo-Jiménez, David Araújo-Vilar. Skipped BSCL2 transcript in Celia's Encephalopathy (PELD): in vitro studies on senescence, adipogenesis and fatty acid treatment. PLOS ONE 2016 Jul 8;11(7):e0158874. doi: 10.1371/journal.pone.0158874.
18. Alejandro Ruiz-Riquelme, Sofía Sánchez-Iglesias, Alberto Rábano, Encarna Guillén-Navarro, Rosario Domingo-Jiménez, Adriana Ramos, Isaac Rosa, Peter Nilsson, Ángel García, David Araújo-Vilar, Jesús R. Requena. Larger aggregates of mutant seipin are part of the molecular basis of Celia's Encephalopathy, a fatal neurodegenerative syndrome associated to the BSCL2 gene. Neurobiology of Diseases, 83:44-53, 2015.
19. Araujo-Vilar D, Sánchez-Iglesias S, Guillín-Amarelle C, Castro A, Lage M, Pazos M, Rial JM, Blasco J, Guillén-Navarro E, Domingo-Jiménez R, Del Campo MR, González-Méndez B, Casanueva FF. Recombinant human leptin treatment in genetic lipodystrophic syndromes: the long-term Spanish experience. Endocrine; 49(1):139-47, 2015.
20. Lado-Abeal J. Thyroid hormones are needed to sustain "inappropriately" normal TSH during nonthyroidal illness syndrome: a clinical observation in severely ill patients with primary hypothyroidism. Neuroendocrinology Letters. 36 (1):101-107. 2015
21. Aráujo-Vilar D, Guillín-Amarelle C, Sánchez-Iglesias S, Castro A, Casanueva FF. Therapeutical uses of recombinant human leptin. Endocrinol Pediat,5 (Suppl): 27-42, 2014
22. Guillín-Amarelle C, Sánchez-Iglesias S, Araújo-Vilar D. Uncommon lipodystrophic syndromes. Medicina Clínica (Barc). 20;144(2):80-7, 2015
23. Guillén-Navarro E; Sánchez-Iglesias S, Domingo-Jimenez R; Victoria B; Ruiz-Riquelme A,Rábano A, Loidi L, Beiras A, González-Méndez B, Ramos A, Lopez González V, Ballesta-Martínez MJ, Garrido- Pumar M, Aguiar P, Ruibal A, Requena JR, Araújo-Vilar D. A new seipin-associated neurodegenerative syndrome. Journal of Medical Genetics. 50; 401-409, 2013
24. Castro I, Quisenberry L, Calvo RM, Obregon MJ, Lado-Abeal J. Lipopolysaccharide induced nonthyroidal illness syndrome causes hypothyroidism and conditions for reduced sensitivity to thyroid hormone.Journal of Molecular Endocrinology. 50: 255-266. 2013.
25. David Araújo-Vilar; Berta Victoria Martínez; Blanca González Méndez; Francisco Barreiro Morandeira; Beatriz Fernández Rodríguez; Rubén Cereijo; José Gallego Escuredo; Francesc Villarroya; Alberto Pañeda Menéndez. Histological and molecular features of lipomatous and non lipomatous adipose tissue from subjects with familial partial lipodystrophy due to LMNA mutations. Clinical Endocrinology. 76: 816-824, 2012.
26. Ana Ramos Levi; Angel Diaz Perez; Jose Manuel Cabezas Agricola; Maria Jesus Sobrido; Sergio Piñeiro; David Araújo-Vilar. Axonal neuropathy, long limbs and bumpy tongue: Think of MEN2B. Muscle&Nerve. 2012; 46: 961-964.
27. Joaquín Lado Abeal; R Albero Gamboa; David Araújo-Vilar; O Barca Mallo; Ignacio Bernabeu Morón; MT Calvo; Isabel Castro Piedras; J Martin Calamata; Fernando Palos Paz; Diego Peteiro. Clinical and molecular study of five families with resistance to thyroid hormones. Medicina Clínica (Barc). 2011; 137: 551 - 554.
28. Diego Peteiro-Gonzalez, Beatriz Fernandez-Rodriguez, Jose M Cabezas-Agrícola, David Araújo-Vilar. Severe localized lipoatrophy related to therapy with insulin analogs in type 1a diabetes mellitus. Diabetes Res Clin Pract, 91:e61-3, 2011.
29. Lado-Abeal J, Castro-Piedras I, Palos-Paz F, Labarta-Aizpún JI, Albero-Gamboa R. A Family with congenital hypothyroidism due to a combination of loss-of-function mutations in the TSH receptor (TSHR) and adenylate cyclase-stimulating G alpha-protein (GNAS) genes.
Thyroid.21:103-109. 2011
30. Berta Victoria, Marta Cuervo, José Manuel Cabezas-Agrícola, Blanca González-Méndez, Giovanna Lattanzi, Rosalba Del Coco, Lourdes Loidi, Francisco Barreiro, Carlos Calvo, David Araújo-Vilar. Reduced adipogenic gene expression in fibroblasts from a patient with type 2 congenital generalized lipodystrophy. Diabetic Medicine 27: 1178–1187, 2010
31. Lado-Abeal J, Romero A, Castro-Piedras I, Rodriguez-Perez A, Alvarez-Escudero J. Thyroid hormone receptors are down-regulated in skeletal muscle of patients with non-thyroidal illness syndrome secondary to non-septic shock.Eur J Endocrinol. 163:765-73. 2010.
32. Lado-Abeal J, Castro P, De la Calzada J, Castro-Piedras I, Celestino R, Espadinha C, Soares P, Sobrinho-Simoes M, Cameselle-Teijeiro J. Identification of a Paired Box Gene 8- Peroxisome Proliferator-Activated Receptor γ (PAX8-PPAR γ) rearrangement mosaicism in a patient with an autonomous functioning follicular thyroid carcinoma bearing an activating mutation in the TSH receptor. Endocrine Related Cancer. 17:599-610. 2010.
33. Peteiro D, Lee J, Rodriguez-Fontan J, Castro I, Cameselle-Teijerio JM, Beiras A, Alvarez CV, Hardy DM, Targovnik HM, Arvan P, Lado-Abeal J. New insights into thyroglobulin pathophysiology revealed by the study of a family with congenital goiter. Journal of Clinical Endocrinology and Metabolism. 95:3522-3526. 2010.
34. Joaquin Lado-Abeal, Rosa-Maria Calvo, Berta Victoria, Isabel Castro, Maria Jesus Obregon, David Araújo-Vilar. Regional decrease of subcutaneous adipose tissue in patients with type 2 familial partial lipodystrophy is associated with changes in thyroid hormone metabolism. Thyroid 20:419-24, 2010
35. Parajes S, Loidi L, Reisch N, Dhir V, Rose IT, Hampel R, Quinkler M, Conway GS, Castro-Feijóo L, Araújo-Vilar D, Pombo M, Dominguez F, Williams EL, Cole TR, Kirk JM, Kaminsky E, Rumsby G, Arlt W, Krone N. Functional Consequences of Seven Novel Mutations in the CYP11B1 Gene: Four Mutations Associated with Nonclassic and Three Mutations Causing Classic 11{beta}-Hydroxylase Deficiency. J Clin Endocrinol Metab. 95:779-88, 2010
36. Araújo-Vilar D, Giovanna Lattanzi, Blanca González-Méndez, Ana Teresa Costa-Freitas, Daniel Prieto, Marta Columbaro, Elisabetta Mattioli, Berta Victoria, Noelia Martínez-Sánchez, Alia Ramazanova, Máximo Fraga, Andrés Beiras, Jerónimo Forteza, Lourdes Domínguez-Gerpe, Carlos Calvo, Joaquin Lado-Abeal Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J Medical Genetics 46: 40-8, 2009
37. Lago, R, Iglesias MJ, San-Jose, E, Areal C, Eiras A, Araujo-Vilar D, Lado-Abeal J. Dominguez-Gerpe L. Prevalence and functional analysis of the S107P SNP (rs6647476) of the SLC16A2 gene in the male population of Northwest Spain (Galicia). Clinical Endocrinology 70(4):636-43, 2009
38. Castro-Piedras I, Lima L, Seoane R, Lado-Abeal J. Identification and functional characterization of two novel activating TSH receptors mutants in toxic thyroid follicular adenomas. Thyroid.19: 645-649. 2009.
39. Domínguez-Gerpe L, Araújo-Vilar D. Prematurely Aged Children: Molecular Alterations Leading to Hutchinson-Gilford Progeria and Werner Syndromes. Current Aging Science 1:202-212, 2008
40. Palos-Paz F, Perez-Guerra O, Cameselle-Teijeiro J, Rueda-Chimeno C, Barreiro-Morandeira F, Lado- Abeal J and the Galician Group for the Study of Toxic Multinodular Goitre: Araujo-Vilar D, Argueso R, Barca O, Botana M, Cabezas-Agrícola JM, Catalina P, Dominguez Gerpe L, Fernandez T, Mato A, Nuño A, Penin M, Victoria B. Prevalence of mutations in TSHR, GNAS, PRKAR1A and RAS genes in a large series of toxic thyroid adenomas from Galicia, an iodine deficient area in NW Spain. European J Endocrinol 159: 623-31, 2008
41. Eva Fernández-Rodríguez, Rocío Villar-Taibo, Iria Pinal-Osorio, José Manuel Cabezas-Agrícola, Urbano Anido-Herranz, Alma Prieto, Felipe F Casanueva, David Araujo-Vilar. Severe hypertension and hypokalemia as first clinical manifestations in ectopic Cushing's syndrome. Arquivos Brasileiros de Endocrinologia & Metabologia, 52(6):1066-70, 2008
42. Araújo-Vilar D, Sarmiento LM, Barros N, Ana Costa da Silva-Freitas, Alia Ramazanova, Joaquín Lado- Abeal, Carlos Calvo, F. Palos and Lourdes Dominguez. Resting metabolic rate is reduced in some obese subjects' subpopulations. Obesity and Metabolism 4: 22-27, 2008
43. Araújo-Vilar D, Joaquin Lado-Abeal, Fernando Palos-Paz, Giovanna Lattanzi, Manuel Angel Bandín, Diego Bellido, Lourdes Domínguez-Gerpe, Carlos Calvo, Oscar Pérez, Alia Ramazanova, Noelia Martínez-Sánchez, Berta Victoria, Costa Freitas AT. A new clinical condition associated with a novel mutation in LMNA gene, characterized by partial lipodystrophy, insulin-resistance, aortic stenosis and hypertrophic miocardiopathy. Clinical Endocrinology 69(1):61-8, 2008.
44. Fernando Palos, María ER García-Rendueles, David Araujo-Vilar, Maria Jesús Obregon, Rosa Maria Calvo, Jose Cameselle-Teijeiro, Susana B Bravo, Oscar Perez-Guerra, Lourdes Loidi, Barbara Czarnocka, Paula Alvarez, Samuel Refetoff, Lourdes Dominguez-Gerpe, Clara V Alvarez, Joaquin LadoAbeal. Pendred syndrome in two Galician families: insights into clinical phenotypes through cellular, genetic and molecular studies. J Clin Endocrinol Metab. 93(1):267-77, 2008.
45. Palos-Paz F, Perez-Guerra O, Cameselle-Teijeiro J, Rueda-Chimeno C, Barreiro-Morandeira F, Lado- Abeal J (and the Galician Group for the Study of Toxic Multinodular Goitre). Prevalence of mutations in TSHR, GNAS, PRKAR1A and RAS genes in a large series of toxic thyroid adenomas from Galicia, an iodine deficient area in NW Spain. European Journal of Endocrinology. 159:623-631. 2008.
46. Rodriguez-Perez A, Palos-Paz F, Kaptein E, Visser TJ, Dominguez-Gerpe L, Alvarez-Escudero J, Lado- Abeal J. Identification of Molecular Mechanisms Related to Non-Thyroidal Illness Syndrome in Skeletal Muscle and Adipose Tissue from Patients with Septic Shock. Clinical Endocrinology (Oxf). 68:821-827. 2008.
47. J.F. Ascaso, E. Aguillo, D. Araujo-Vilar et al. Diabetes mellitus y riesgo cardiovascular. Recomendaciones del grupo de trabajo Diabetes mellitus y enfermedad cardiovascular de la Sociedad Española de Diabetes 2006. Clin Invest Arterioscl. 19(3):167-72, 2007
48. Lado-Abeal J, Lorenzo-Solar M, Lago-Lestón R, Palos-Paz F, Domingez-Gerpe L. Hyperglycaemic hyperosmolar non-ketotic state as a cause of low gonadotropin levels in postmenopausal diabetic women: a role for severe hypernatraemia. Journal of Neuroendocrinology. 19:983-987. 2007
49. Villamil Cajoto I, Araujo-Vilar, D. Tratamiento con vitamina D en la infancia: discusión de la evidencia. An. Med. Int. 23:446-48, 2006
50. Araujo-Vilar D, Grupo De Trabajo Diabetes Mellitus y Enfermedad cardiovascular. Documento de consenso: Diabetes mellitus y riesgo cardiovascular. Av. Diabetol 22: 143-48, 2006
51. Loidi L, Quinteiro C, Parajes S, Barreiro J, Leston DG, Cabezas-Agricola JM, Sueiro AM, Araujo-Vilar D, Castro-Feijoo L, Costas J, Pombo M, Dominguez F. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf). 2006 Mar;64(3):330-6, 2006.
52. Dumitrescu AD, Liao XH, Abdullah SYM, Lado-Abeal J, AbdulMajed F, Moeller LC, Boran G, Schomburg L, Weiss RE, Refetoff S. Mutations in the selenocystein insertion sequence binding protein (SBP)2 produce a thyroid phenotype. Nature Genetics. 37:1247-1252. 2005
53. Lado-Abeal J, Dumitrescu AM, Cohen RN, Pohlenz J, Liao XH, Lebrethon MC, Verloes A, Refetoff S.A de novo mutation in an Already Mutant Nucleotide of the Thyroid Hormone Receptor Beta Gene perpetuates resistance to thyroid hormone. Journal of Clinical Endocrinology and Metabolism. 90:1760-1767. 2005.
54. Lado-Abeal J, Robert-McComb JJ, Qian XP, Leproult R, Van Cauter E, Norman RL. Gender differences in the neuroendocrine response to short-term energy restriction in non-human primates. Journal of Neuroendocrinology. 17:435-44. 2005.
55. Graña Barcia M, Liz Leston JL, Lado Abeal J. Subcutaneous administration of pulsatile GnRH decreases serum FSH and LH levels in women with polycystic ovary syndrome: a preliminary study. Fertility and Sterility. 83:1466-1472. 2005
56. García-Estevez DA, Araujo-Vilar D, Saavedra-Gonzalez A, Fiestras-Janeiro G, Cabezas-Cerrato J. Analysis of the relationship between body mass index, insulin resistance, and beta-cell function: a crosssectional study using the minimal model. Metabolism. 53(11):1462-6, 2004.
57. Lado-Abeal J, DeValk E, Pacini F, Refetoff S. Study of the effect of recombinant human thyroid stimulating hormone on Leydig cells function in men with differentiated thyroid carcinoma. Thyroid. 13:649-652. 2003.
58. Narendran P, Lado-Abeal J, Moeller LC, Refetoff S. Partial thyroxine-binding globulin (TBG) deficiency in a family with no detectable mutation of the TBG gene. Clinical Endocrinology. 59:823-825. 2003.
59. Cabezas-Cerrato, Araújo-Vilar D. Resistencia a la acción de la insulina. Evolución histórica del concepto. Técnicas para el estudio in vivo en humanos. Endocrinología y Nutrición. 50: 396-406, 2003
60. Garcia-Estevez DA, Araujo-Vilar D, Fiestras-Janeiro G, Saavedra-Gonzalez A, Cabezas-Cerrato J. Insulin resistance in essential hypertension: a conflictive point of view. Diabet Med. 2003 Dec;20(12): 1035, 2003.
61. Araujo-Vilar D, Loidi L, Dominguez F, Cabezas-Cerrato J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm Metab Res. 2003 Jan;35(1):29-35, 2003.
62. Garcia-Estevez DA, Araujo-Vilar D, Fiestras-Janeiro G, Saavedra-Gonzalez A, Cabezas-Cerrato J. Comparison of several insulin sensitivity indices derived from basal plasma insulin and glucose levels with minimal model indices. Horm Metab Res. 35(1):13-7, 2003.
63. Araújo-Vilar D. Lipodystrophies: molecular basis and clinical characteristics. Endocrinology. 50: 133-44, 2003
Articles in preparation:
1. David Araújo-Vilar, Ferruccio Santini. Diagnosis and treatment of lipodystrophies: a step-by-step approach.
2. D Araujo-Vilar et al. A case of Celia's encephalopathy due to a new BSCL2 mutation.
3. S. Sánchez-Iglesias, D Araujo-Vilar et al. Role of BSCL2 transcripts in human brain and its relationships with ROS protection and peroxisomes generation.
4. Cristina Guillín-Amarelle, Sofía Sánchez-Iglesias, David Araújo-Vilar. VARIABLE EXPRESSIVITY IN LMNA-ASSOCIATED FAMILIAL PARTIAL LIPODYSTROPHY.
5. Sofía Sánchez-Iglesias, Marcos M. Lima-Martínez, Cristina Guillín-Amarelle, S, David Araújo-Vilar. LEVOTHYROXINE TREATMENT IN RABSON-MENDENHALL SYNDROME.
6. C Guillín-Amarelle, S Sánchez-Iglesias, A Fernández-Pombo, D Araujo-Vilar. How to diagnose a LMNAassociate lipodystrophy. Nucleus 2017
Organizing conferences and other scientific activities
Meeting of the European Consortium Lipodystrophies:
• Bologna, 10 to 11 February 2014 (Prof. Giovanna Lattanzi)-Birth founding. (figure 15)

• Paris, April 30, 2015 (Prof. Corinne Vigouroux) (Figure 16)

• Santiago de Compostela, 8 to 9 March 2016 (Prof. David Araujo-Vilar) (Figure 17)

• Rome, 21 to 22 April 2017 (Prof. Giuseppe Novelli) (Figure 18, 19, 20)



• Ulm, 7 to 8 July 2017 (Prof. Martin Wabitsch)
• Münster, 27 to 28 April 2018 (Prof. Hartmut Schmidt)
Collaborations with other research groups:
• Dra Rosario Domingo, Unit Neuropediatría, HospitalUniversitario Virgen de la Arrixaca, Murcia
• Prof. Giovanna Lattanzi, National Research Council of Italy – Institute for Molecular Genetics, Bologna, Italy.
• Prof. Corinne Vigouroux, Center de Recherche Saint-Antoine INSERM UMR_S 938. Faculté de Médecine Pierre et Marie Curie (Figure 21) .

• Dr. David Savage, National Severe Insulin Resistance Service. Wellcome Trust-MRC Institute of Metabolic Science. Cambridge University Hospital NHS Foundation Trust . Addenbrookes Hospital. UK (Figure 22).

• Prof. Steve Young, University of California, Los Ángeles, EEUU.
• Dra. Rebecca Brown, National Institute of Diabetes and Digestive and Kidney, Diseases( NIDDK), Diabetes, Endocrinology, and Obesity Branch (DEOB), Bethesda, MD, EEUU.
• Prof. Francesc Villarroya, Department of Biochemistry and Molecular Biology, Universitat de Barcelona (Figure 23).

• Prof. Martin Wabitsch, Pediatrics and Adolescent Medicine, University Medical Center Ulm, Alemania.
• Dra. Margarita Lopez Trascasa, Immunology, University Hospital La Paz, Madrid.
• Prof. Elif A. Oral, University of Michigan, EEUU (Figure 24)

Contact
1. For matters related to research projects:
Prof. David Araujo-Vilar
UETeM, Lab 3, Floor 2
CIMUS, Avda Barcelona 3
15707 Santiago de Compostela
Tfn: +34 639 393 458
Mail: david.araujo@usc.es; lipodistrofias@gmail.com
2. For medical issues:
Prof. David Araujo-Vilar
Endocrinology and Nutrition University Hospital
Complex of Santiago.
(Hospital Medical-Cirúrxico Conxo, Rua Ramón Baltar s/n, 15782 Santiago)
Mail: david.araujo@usc.es; lipodistrofias@gmail.com
Note: Health care in a public hospital is free. As our service unit of reference in Spain for Lipodystrophies, it is recommended that patients who do not belong to the Galician Health Service (SERGAS) apply to your family doctor or specialist who request healthcare through the SIFCO system (Information System Cohesion Fund) so they can be seen at our service.